Medical devices on the market are under strict surveillance by governing bodies such as the FDA. As with all tech nowadays, the Medical Device industry is evolving at hyperspeed, and regulations need to constantly keep up to date in order to continue to save lives. Patient safety is of paramount importance, and risk-based classification of Medical Devices lies at the core of upholding that. Since the industry is constantly in flux, classification and reclassification of medical devices is a never-ending process.
How the FDA classifies medical devices
Classifying medical devices is a thorough process. The FDA categorizes medical devices into one of three classes – Class I, II, or III – based on their risks and the regulatory controls necessary to provide a ‘reasonable assurance of safety and effectiveness’, with Class I indicating lowest risk and least controls required, and Class III the highest with the most controls. Risk is assessed based on the invasiveness of the device and its potential impact on the overall health of the patient.
As of today, The FDA has classified around 1,700 different generic types of devices, grouped into 16 medical panels as defined by 21 CFR 862 to 21 CFR 892. These devices are then assigned to one of the three regulatory classes. As well as being risk-based, FDA device classification depends on the intended use of the device and also upon indications for use. For example, a scalpel's intended use is to cut tissue. A more specialized indication may be added in the device's labeling or may be given verbally during product sale, in this example this indication could be "for making incisions in the cornea". For more about the meaning of intended use, this FDA article is useful.
Breaking down Class I, II, and III medical devices
1. Class I Medical Devices
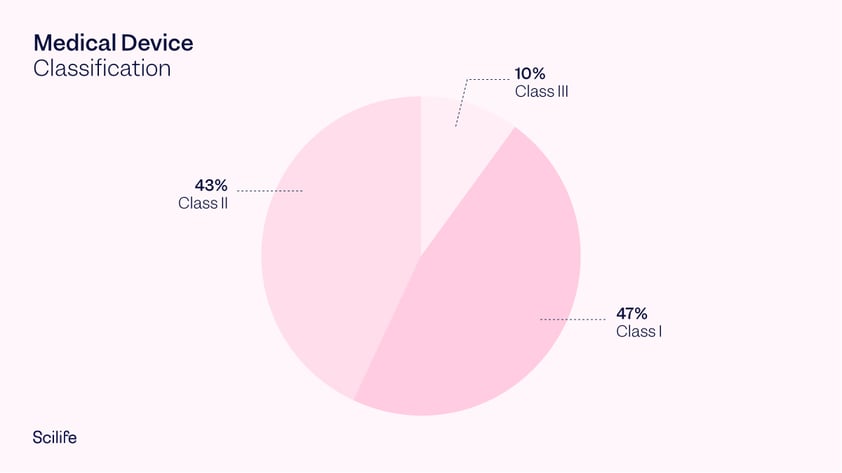
Class I medical devices are those that have a low to moderate risk to the user. Given the low risk involved, these devices are only required to meet general controls. This is the reason why 47% of medical devices fall under this category and 95% of these are exempt from the regulatory process. If a device falls into a generic category of exempted Class I devices, a premarket notification application and FDA clearance is not required before marketing the device in the U.S. However, the manufacturer is required to register their establishment and list their generic product with the FDA. Examples of Class I medical devices include wheelchairs, elastic bandages, manual stethoscopes, and bedpans.
2. Class II Medical Devices
Class II medical devices are those with a moderate to high risk to the user. These are the devices which require general as well as special controls. Today, 43% of medical devices fall under this category. Examples of Class II devices include catheters, needles and contact lenses.
3. Class III Medical Devices
Class III medical devices carry a high risk to the patient. These devices need to meet many general controls and obtain pre-market approval. They are usually devices that sustain or support life, are implanted, or present a potentially unreasonable risk of illness or injury. Class III medical devices represent 10% of medical devices regulated by the FDA. Examples include implantable pacemakers, coronary stents, orthopedic implants and breast implants.
How to find out the class of your medical device
The FDA provides excellent resources for determining classification and whether exemptions may exist. There are various ways to find out what class your medical device falls under, including:
- The most common method is to search the FDA classification database. Search for a part of the device name, or, if you know the device panel (medical specialty) to which your device belongs, go directly to the listing for that panel and identify your device and the corresponding regulation.
- Search for a similar device by surfing the 501(k) Clearance Database, the Pre-market Approval Database or the De Novo Database.
- Search for a similar device by surfing the Establishment Registration and Device Listing Database.
What controls must medical devices meet?
1. General Controls
General Controls which all Class I, II and III medical devices need to meet include the provisions of the Act pertaining to:
-
- Adulteration;
- Misbranding;
- Device registration and listing;
- Premarket notification;
- Banned devices;
- Notification and repair, replacement, and refund;
- Records and reports;
- Restricted devices; and
- Good Manufacturing Practices.
- Adulteration;
2. Special Controls
Additional FDA ‘Special Controls’ are applicable to class II devices and include:
-
- Special Labeling
- Design Characteristics or Specifications
- Performance Standards
- Premarket Data Requirements
- Guidance Documents
- Special Labeling
3. Premarket Approval
This is typically a requisite for life supporting and life sustaining Class III devices, since General Controls and Special Controls are insufficient to provide reasonable assurance of safety and effectiveness in this case.
Getting your medical device to market, fast
Owing to their high risk potential, Class III medical devices are required to meet much tighter standards of safety and efficacy compared to Class I and Class II devices. Most Class III medical devices need to undergo Premarket Notification 510(k) and Premarket Approval to prove substantial equivalence as per 21 CFR 814, whereas Class I and Class II devices are usually exempt from this process. This means Class I and Class II medical devices can enter into the U.S. market much faster.
Scilife offers intuitive modules that can accelerate your medical device to market, by streamlining the processes required for regulatory approval.
Discover Scilife for Medical Devices